Qu'est ce que l'OI ?
La pathologie
OI est l’abréviation de « ostéogénèse imparfaite », littéralement « formation imparfaite des os ». L’ostéogenèse imparfaite est une maladie génétique rare due à un défaut du collagène, élément principal de l’os. Elle se caractérise par une fragilité osseuse, c’est pourquoi elle est plus couramment connue sous le nom de « maladie des os de verre ».
C’est une affection génétique qui peut être transmise par l’hérédité ou résulter d’une mutation. En France, 50 à 60 enfants atteints d’Ostéogenèse Imparfaite naissent chaque année (1 naissance sur 10 000). Elle se présente sous des formes de gravité très variable.
 |
 |
_________________________________________________________________________
Les conséquences
Les conséquences de l’OI peuvent être : des fractures fréquentes, parfois même en l’absence de tout traumatisme, des déformations osseuses, une petite taille, des douleurs chroniques, une fragilité dentaire, une hyperlaxité ligamentaire, une surdité plus ou moins sévère à l’âge adulte, des hématomes spontanés, une fatigabilité.
Chez certains, la maladie peut ne pas être apparente, tandis que d’autres personnes atteintes sont de petite taille et présentent des déformations du squelette.
Dans tous les cas, une prise en charge pluridisciplinaire précoce et continue aidera les personnes atteintes à mener une vie la plus autonome possible.
_________________________________________________________________________
Les mécanismes à l’origine de l’OI
Par Dominique Duménil et Françoise Moreau-Gachelin, de l’association ScienS’As, et Marie-Christine de Vernejoul, ancienne Présidente du Conseil scientifique de l’AOI
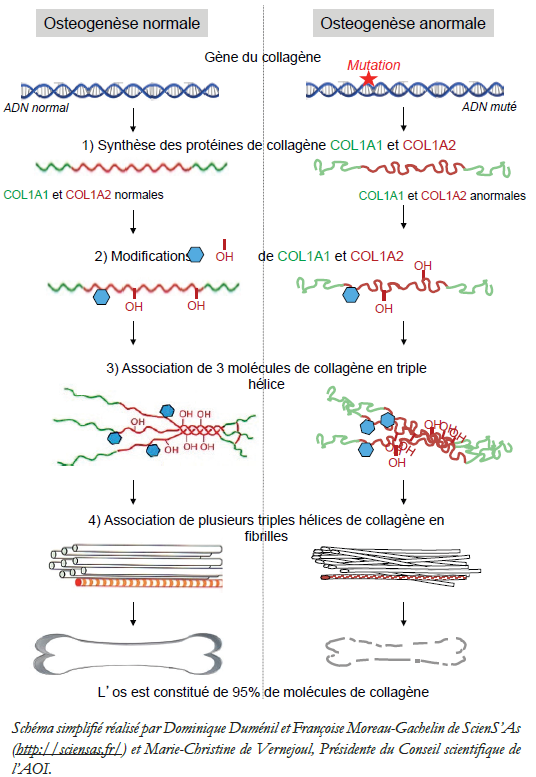
L’os est constitué de 95% de collagène. Les molécules de collagène 1 (COL1A1 et A2) codées par les gènes du collagène (fig.1) sont synthétisées par une cellule de l’os : l’ostéoblaste. Au cours du développement, ces molécules subissent de nombreuses modifications biochimiques (fig.2) avant d’être associées en triple hélice (fig.3), puis transportées dans la matrice osseuse où elles forment des petites fibres appelées fibrilles (fig.4). Ces étapes sont régulées par différentes protéines.
Si des modifications génétiques affectent soit la molécule de collagène, soit une des molécules impliquées dans les différentes étapes de la transformation de ce collagène, les fibrilles ne sont pas normales ce qui entraîne une fragilité de l’os et des fractures.
Chez environ 90% des patients, la mutation génétique porte sur le gène qui code le collagène 1. La gravité de la maladie dépend de l’endroit de la mutation sur le gène. Le siège de la mutation est différent dans chaque famille.
Récemment, des modifications génétiques d’autres gènes que le collagène 1 ont été caractérisées chez des patients. Des mutations dans les gènes codant les protéines qui régulent la formation des fibres de collagène et la formation osseuse (CRTAP, LEPRE1, PPIB, SERPINH1, SERPINF1, SP7…) ont été décrites. Les cliniciens/chercheurs ont tenté de corréler les symptômes à ces modifications génétiques et montrent que les désordres observés chez les patients dépendent, en partie, de la nature de la modification génétique.
_________________________________________________________________________
Classification de l’OI
Les différentes formes d’OI ont été classées en différents types cliniques en fonction de leur sévérité. Au sein de chacun des types, l’OI peut néanmoins être causée par différentes mutations génétiques. Jusqu’à récemment, un nouveau type était ajouté à la classification chaque fois qu’un nouveau gène responsable de l’OI était découvert (OI de type VI, etc.). Mais depuis 2015/2016, l’OI est communément classée en 5 types basés sur la forme clinique et la sévérité, et pour chacun des types sont précisés les mutations spécifiques et le mode de transmission.
Même si la mutation génétique n’est pas identifiée, l’OI peut être diagnostiquée sur la base de la clinique. Il y a encore des mutations responsables de l’OI qui n’ont pas été trouvées ou décrites.
| Nom de la forme d’OI | Type clinique | Mutations génétiques associées | Mode de transmission |
|---|---|---|---|
| Non déformante avec sclérotiques bleues | I | COL1A1, COL1A2 | Dominant |
| OI commune, variable avec sclérotiques normales | IV | COL1A1, COL1A2 WNT1 CRTAP, PPIB PLS3 |
Dominant Dominant Récessif Lié à l’X |
| OI avec calcification des membranes | V | IFITM5 | Dominant |
| OI déformante | III | COL1A1, COL1A2 BMP1, CRTAP, FKBP10 LEPRE1, PLOD2, PPIB SERPINF1, SERPINH1 TMEM38, WNT1 CREB3L1 |
Dominant Récessif Récessif Récessif Récessif Récessif |
| OI létale en périnatal | II | COL1A1, COL1A2 CRTAP, LEPRE1, PPIB |
Dominant Récessif |
_________________________________________________________________________
Diagnostic
L’ostéogenèse imparfaite (OI) est un groupe de maladies génétiques caractérisées par une fragilité osseuse, une faible masse osseuse et une tendance aux fractures de sévérité variable. Elle se présente sous différentes formes dont les caractéristiques ont permis un classement en différents types.
Le diagnostic peut être suspecté en anténatal sur l’échographie et/ou confirmé par l’analyse génétique si la pathologie a déjà été identifiée dans la famille. Lorsqu’aucun antécédent familial n’existe, la maladie peut se révéler par de nombreuses fractures et des malformations osseuses de l’enfant déjà in utero, ou plus tard par des fractures répétées dans la petite enfance.
Pour plus d’informations, consultez le site Orphanet, portail Internet des maladies rares et des médicaments orphelins.
_________________________________________________________________________
Pronostic
Le pronostic fonctionnel est lié à la sévérité de l’OI, à la qualité de soin et sa prise en charge. L’espérance de vie des patients OI n’est pas différente de celle de la population générale, sauf pour certaines formes sévères car alors, le pronostic vital dépend des complications respiratoires associées aux déformations de la colonne vertébrale provoquées par la fragilité osseuse.
_________________________________________________________________________
Une prise en charge multidisciplinaire
La prise en charge doit être multidisciplinaire, par des spécialistes expérimentés en médecine, orthopédie, kinésithérapie et rééducation. Les bisphosphonates, puissants inhibiteurs de la résorption osseuse, sont aujourd’hui le traitement de choix des formes sévères, mais ne sont pas curatifs. La prévention du déficit en vitamine D et calcium est fondamentale à tout âge. La chirurgie est essentielle pour corriger les déformations osseuses et rachidiennes et prévenir les fractures des os longs (enclouage centromédullaire : tige de métal insérée dans la moelle de l’os). La kinésithérapie précoce améliore l’autonomie en facilitant l’évaluation des déficits moteurs, réduisant le risque de chutes et encourageant les patients à la pratique sportive.
Pour plus d’informations, consulter la fiche de synthèse du site Orphanet qui détaille les différents traitements possibles
_________________________________________________________________________
Ressources
Podcasts OI :
![]() Podcast vivre avec OI > Podcast : 3 - Vivre avec une Ostéogenèse Imparfaite
Podcast vivre avec OI > Podcast : 3 - Vivre avec une Ostéogenèse Imparfaite
![]() Podcast qu’appelle t’on OI ? > https://www.aoi.asso.fr/actualite/podcast-1-qu-appelle-t-on-ostogense-imparfaite-25.html
Podcast qu’appelle t’on OI ? > https://www.aoi.asso.fr/actualite/podcast-1-qu-appelle-t-on-ostogense-imparfaite-25.html
Portraits OI :
![]() Portraits de Cassandra & Malya : vivre avec l'OI > https://youtu.be/alRM0FITi00
Portraits de Cassandra & Malya : vivre avec l'OI > https://youtu.be/alRM0FITi00
![]() Ma vie avec l'Ostéogenèse Imparfaite, portrait de Joël > https://youtu.be/blfz4ZBGUOo
Ma vie avec l'Ostéogenèse Imparfaite, portrait de Joël > https://youtu.be/blfz4ZBGUOo
![]() Portraits d'Agnès et Paul - part1&2 > https://youtu.be/uahCr_DSLuE
Portraits d'Agnès et Paul - part1&2 > https://youtu.be/uahCr_DSLuE
> https://youtu.be/SwybiGzuZ04
![]() Portrait de Marie > https://youtu.be/uUNMXtDXSa4
Portrait de Marie > https://youtu.be/uUNMXtDXSa4
